
Prepare data
Convert h5 format to cbf format: XDS can't operate with h5 format images, so you need to convert the image to cbf format before the process. On macOS, eiger2cbf is a useful program to do this.
eiger2cbf path/to/your/h5_master_data -- get number of frames
eiger2cbf path/to/your/h5_master_data N out.cbf -- write N-th frame to out.cbf
eiger2cbf path/to/your/h5_master_data N -- write N-th frame to STDOUT
eiger2cbf path/to/your/h5_master_data N:M out -- write N to M-th frames to outNNNNNN.cbf
For example:
eiger2cbf path/to/your/h5_master_data 1:720 out # convert all the 720 images to cbf format
X-Ray data processing
Process with XDS
XDSGUI
-
Install XDSGUI according to this instrucment.
-
Run the XDSGUI in the terminal.
cd path/to/your/images xdsgui -
Load the image and generate the XDS.INP file:
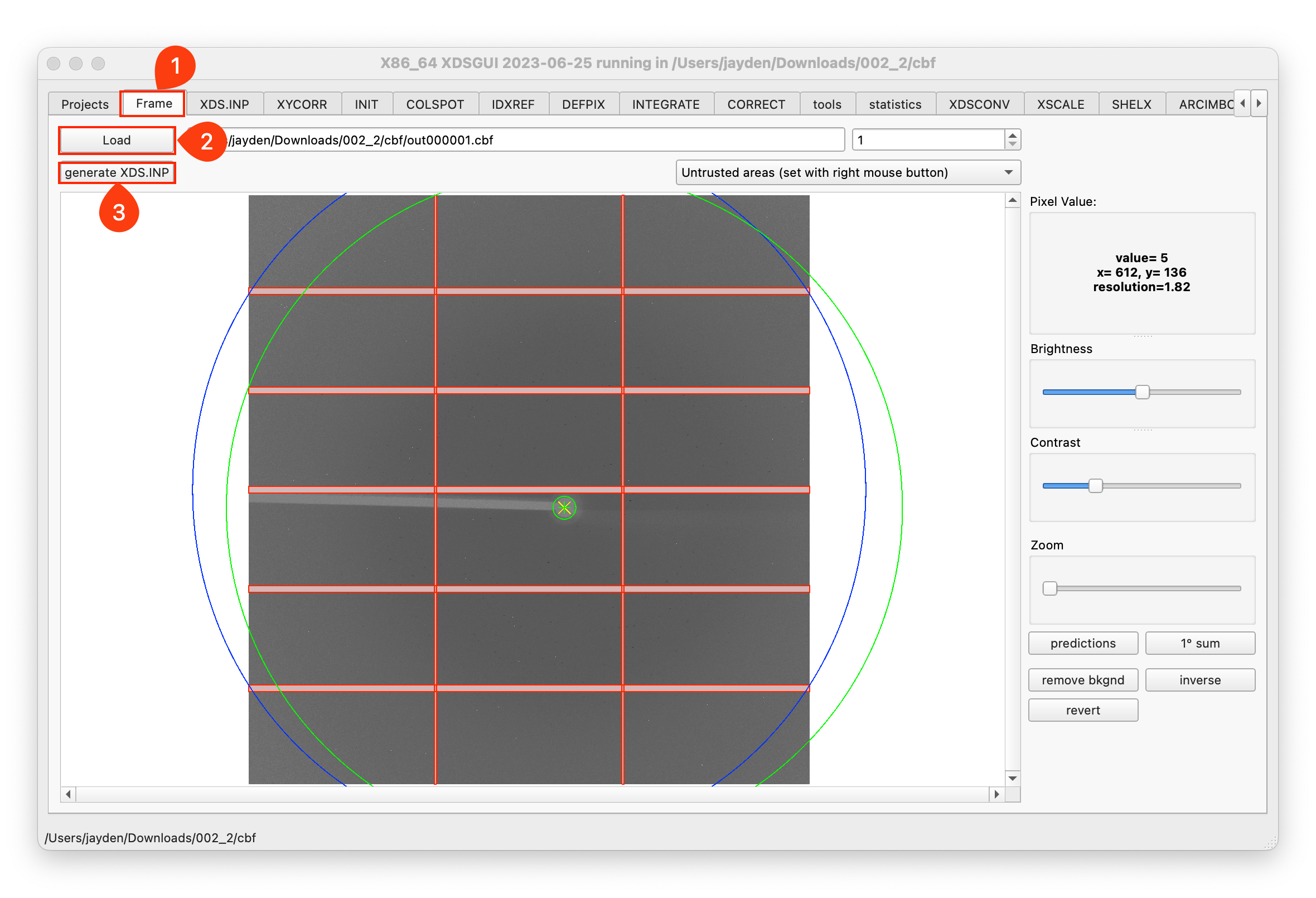
-
Modify the XDS.INP file to meet your needs, then click
Run XDS.:::caution
If you collect your data with 10U2 beamline in SSRF, you should modify the
ROTATION_AXISparameter to0 -1 0.:::
-
Check the result in page CORRECT
Process with Xia2
-
There are two ways to use Xia2: use Xia2 in CCP4, or install the latest xia2/DIALS bundle according to this website.
-
Process your data with it:
cd path/to/images xia2 pipeline=dials-aimless ./ goniometer.axes=0.000000,1.000000,0.000000 xia2.settings.resolution.d_min=2
Process with autoPROC
-
Request a license and install autoPORC according to this website.
-
Process your data with it:
cd path/to/images process -I . -d autoproc -R 50 2 > out.log #save the result in subdirectory named autoproc, resolution vary from 2 to 50, save the log to out.log file.
Structure determination
Valid the data
-
Find the
XDS_ASCII.HKLfile in the autoproc directory as the diffraction file. -
Valid the data with Phenix Xtriage, put the diffraction file in, and run.
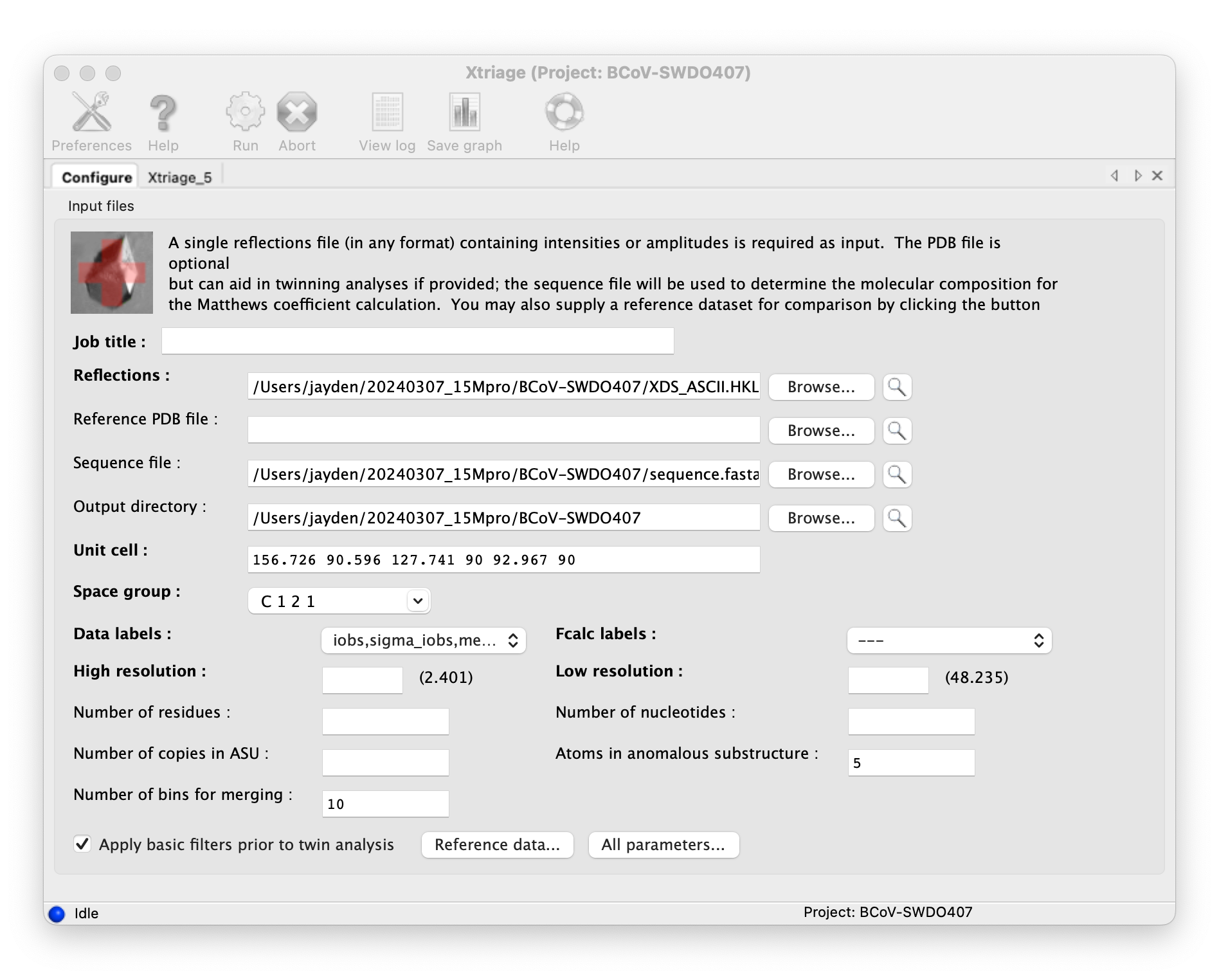
Molecular replacement
-
Find the
XDS_ASCII.HKLfile in the autoproc directory as the diffraction file. -
Get the template model:
- Blast the sequence of your protein with the PDB database, select the proper structure, and download the PDB file.
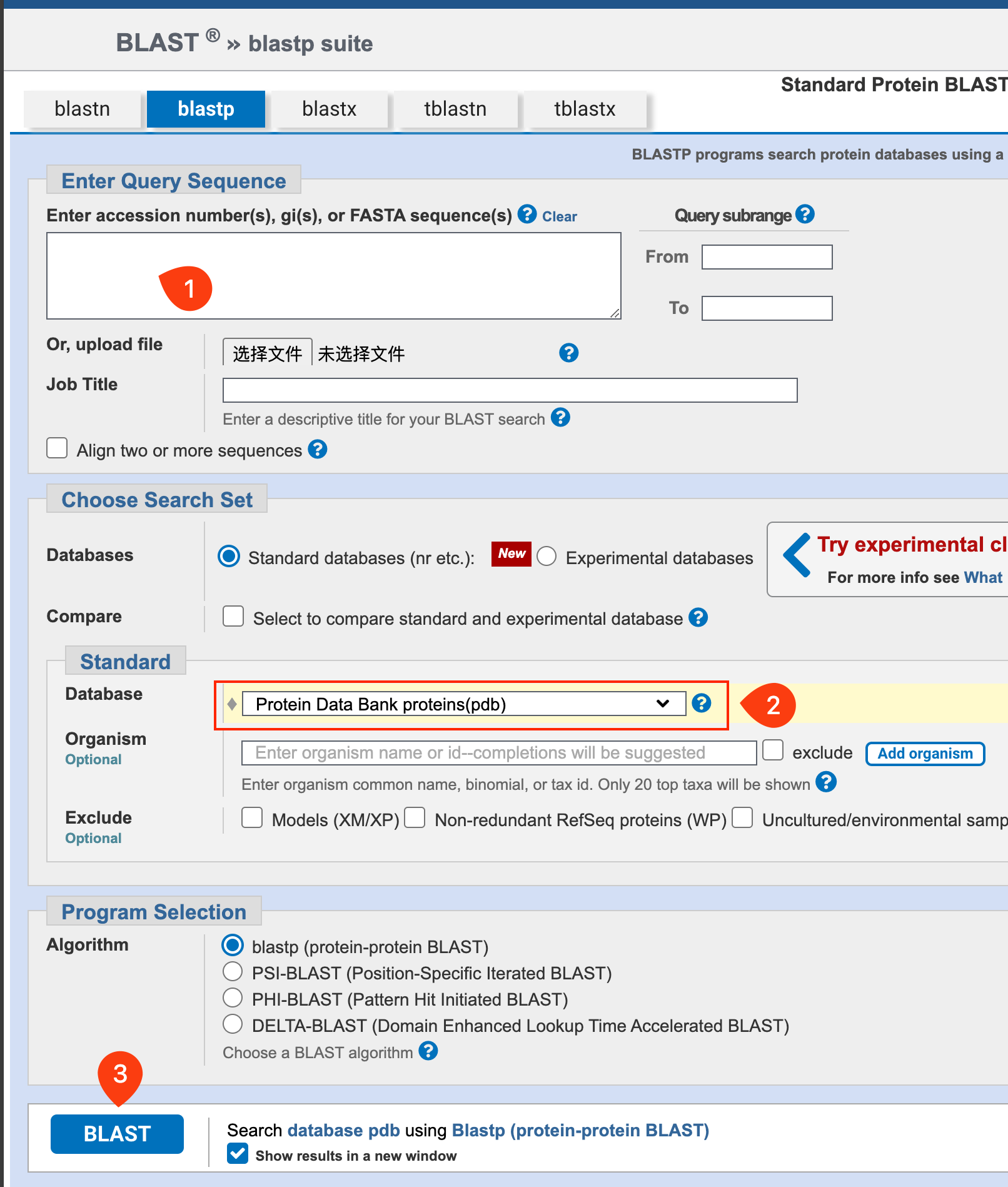
- After downloading the template model, you need to edit it with PyMOL to remove the redundant copies and water molecules.
-
Determine how many copies are in ASU(asymmetric unit):
-
You can get the number from the result of the Xtriage
::: grid {cols=2,rows=1,gap=12,type=images}

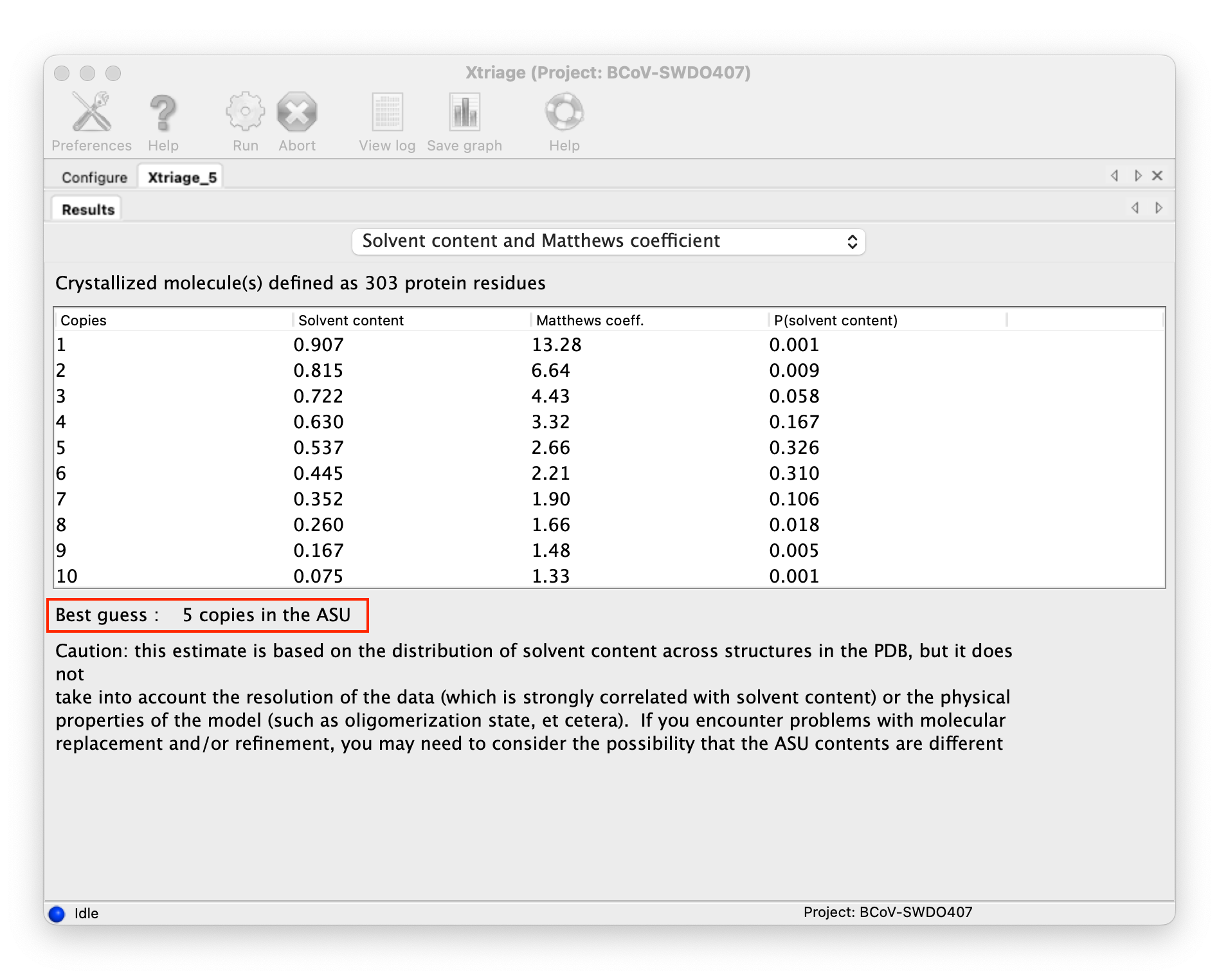
::: -
Or you can use the cell content analysis tool in CCP4.
[!NOTE]
You can determine the number of copies through the Solvent content, generally, the solvent content of most biological macromolecule crystals is between 40% and 60%.
-
-
Do molecular replacement with Phenix Phaser-MR.
::: grid {cols=4,rows=1,gap=12,type=images}
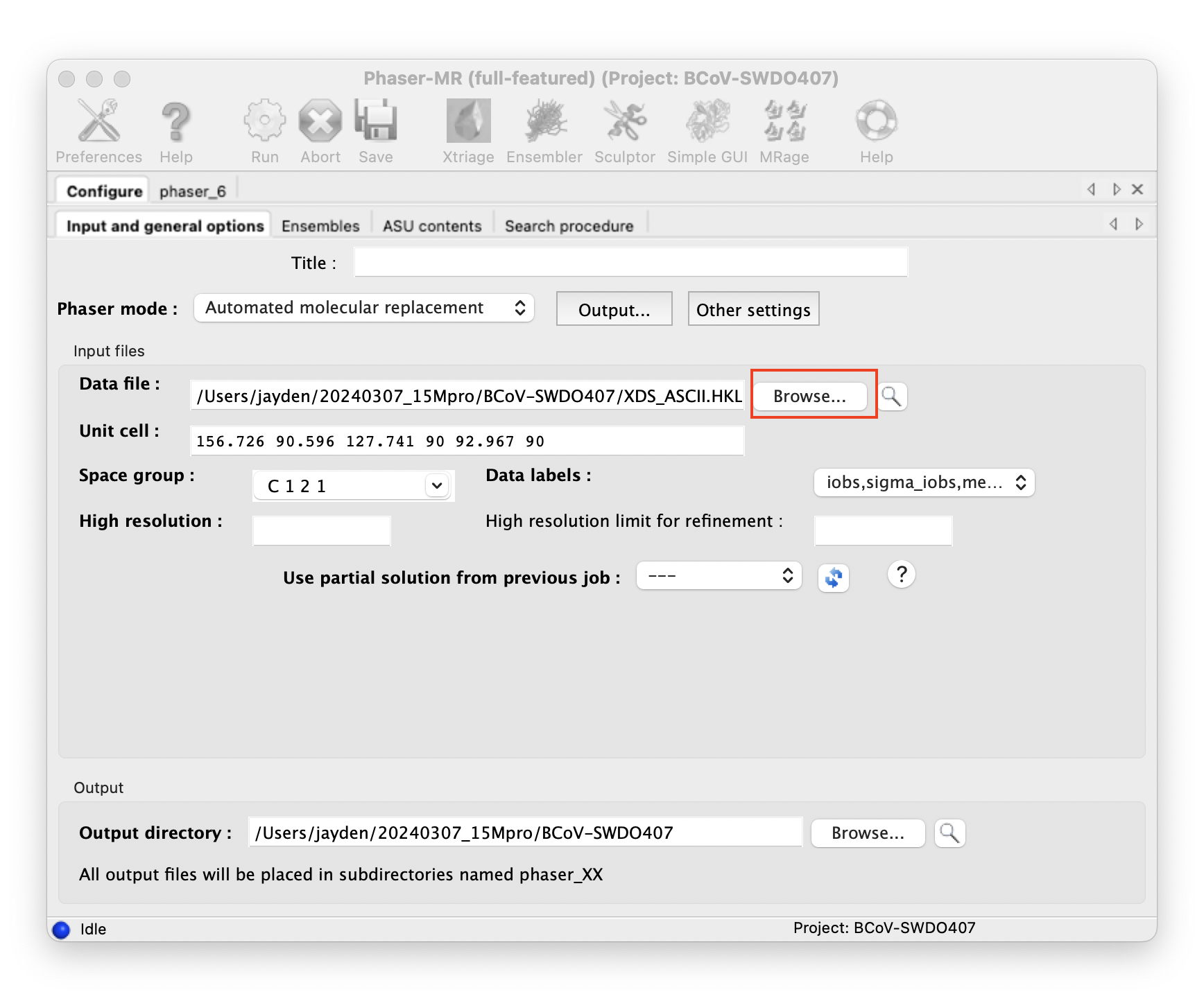
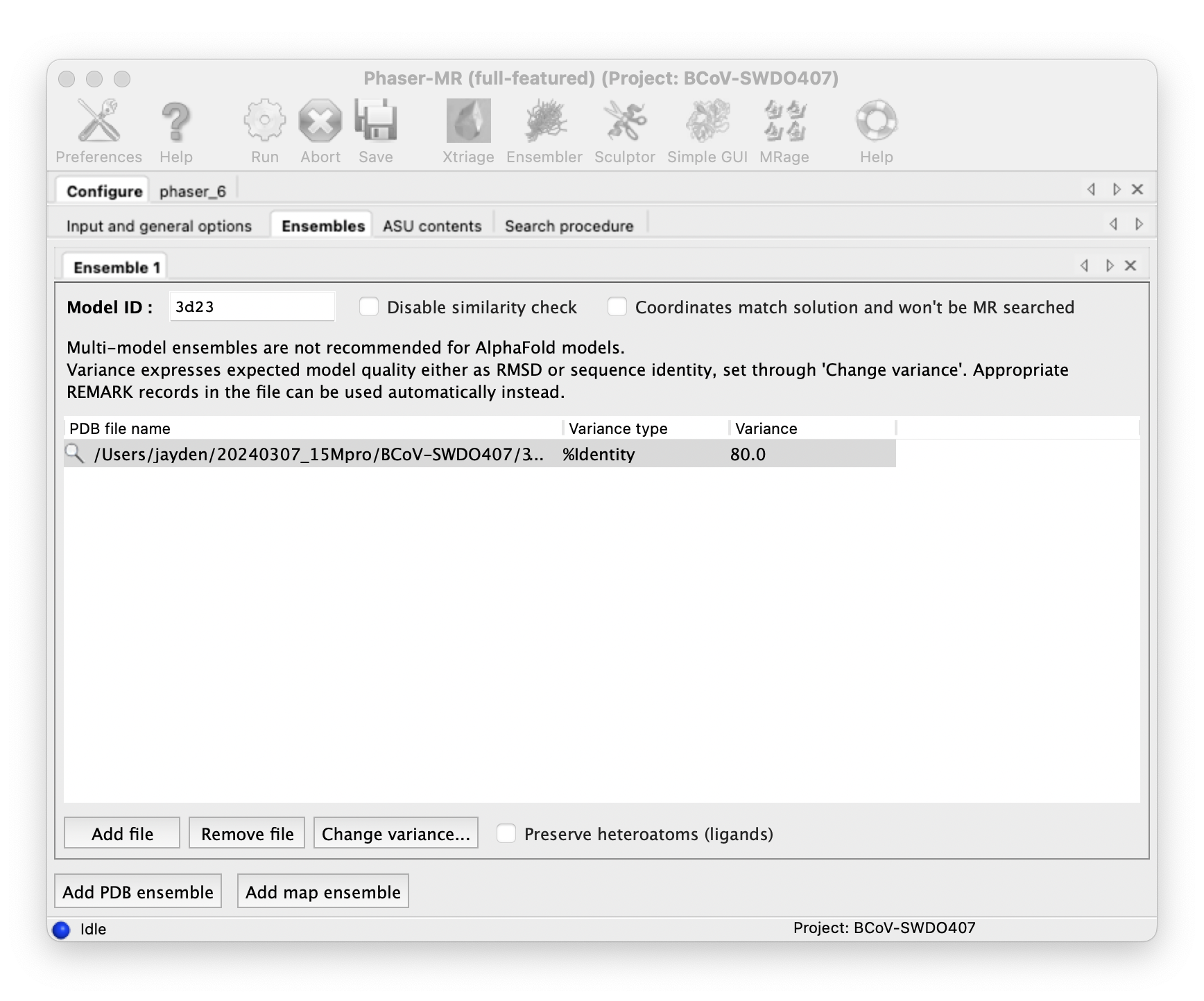
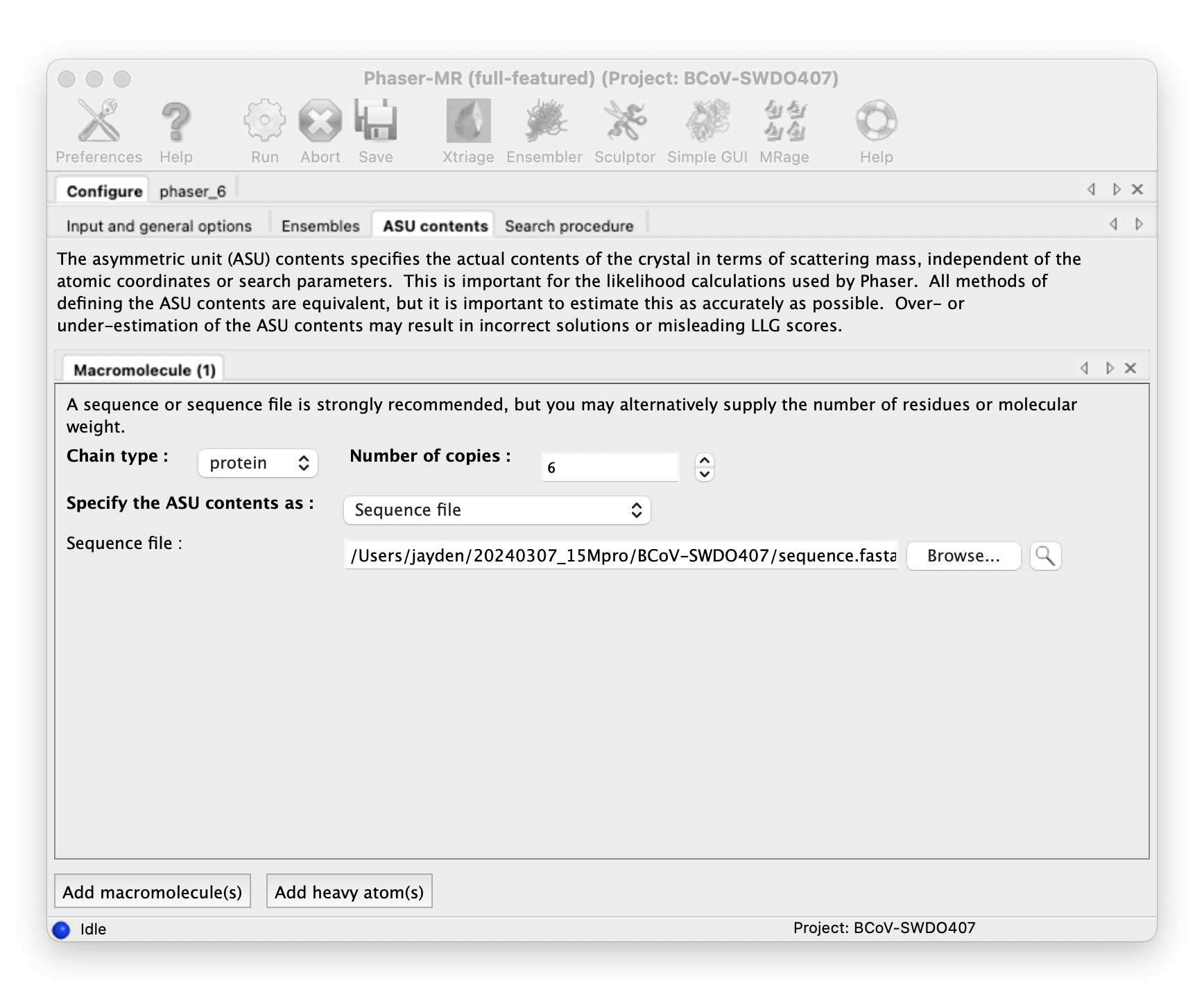
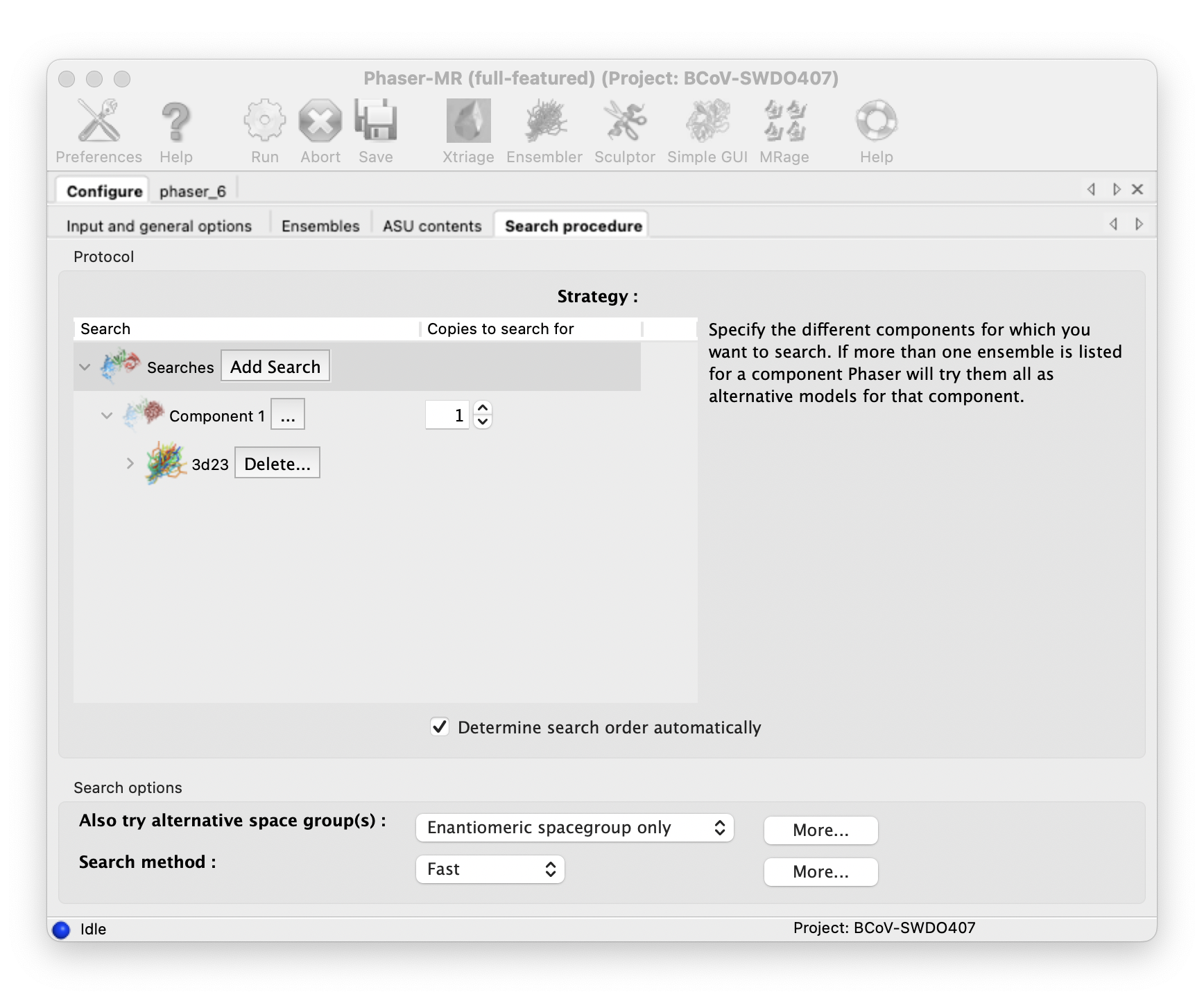
::: -
Build your model with Phenix AutoBuild: input the result of molecular replacement, and run.
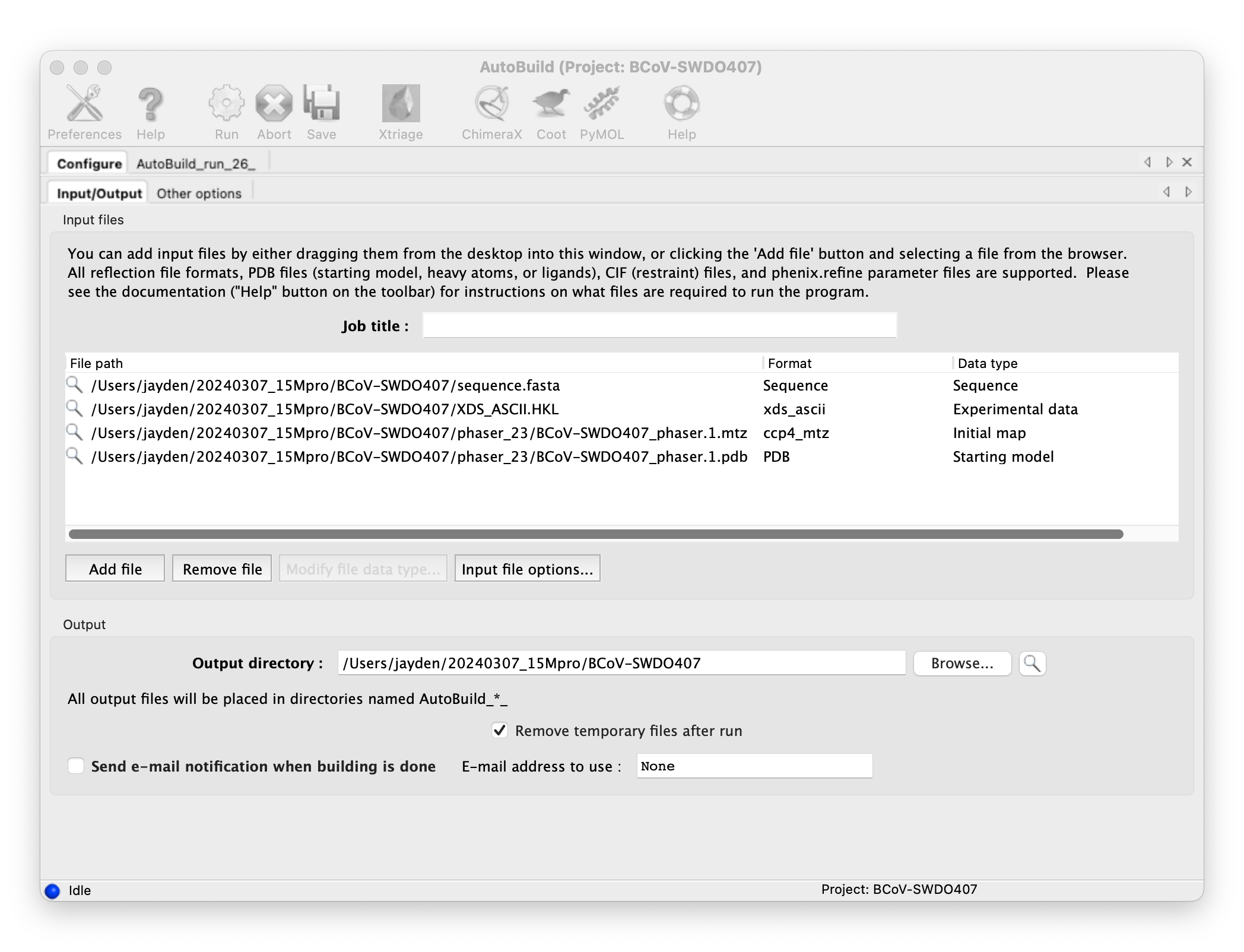
-
Check the model from AutoBuild in Coot and refine it manually.
::: grid {cols=2,rows=1,gap=12,type=images}


:::
此文由 Mix Space 同步更新至 xLog
原始链接为 https://xxu.do/posts/academic/X-ray-data-processing